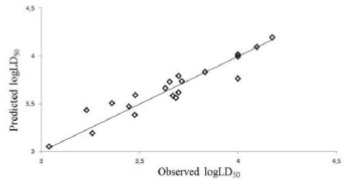
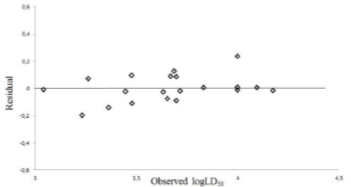
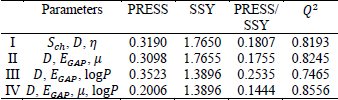
Abstract-- Sulfonamides are one of the most important classes of chemicals found in the aquatic environment, as a pollutant due to excessive consumption. The DFT- B3LYP method with the basis set 6-311++G (d,p), was employed to calculate various quantum chemical descriptors of sulfonamide molecules. A quantitative structure activity relationship (QSAR) study was performed for the toxicity value LD50 of sulfonamides, with their quantum chemical descriptors, by multi linear regression. The QSAR models were validated by internally and externally. The best multilinear equation with correlation coefficient, R, and the cross-validation leave-one-out correlation coefficient, Q2, values were 0.9528 and 0.8556, respectively The results show that the QSAR models have both favourable estimation stability and good prediction power.
Keywords-- DFT, sulfonamides, quantum chemical descriptors, multi linear regression.
I. INTRODUCTION
Sulfonamides (SAs), one of the most important classes of chemicals, are widely used in aquaculture, livestock husbandry and human medicine (Li et al., 2016). They have been used in high volumes for several decades because of their effectiveness and inexpensiveness (Chandran et al., 2011). As a consequence of excessive consumption of SAs, they are frequently detected in the aquatic environment and accumulated the in the food chain (Qin et al., 2016; Lu et al., 2015; Yang et al., 2010; Shah et al., 2015; Voigt et al., 2017). This problem has become important due to the potential toxicity for human health and all living organisms (Mondal et al., 2015; Guo et al., 2012).
Testing toxicity is often restricted by its high cost, time consuming experimental procedures, public objection to animal testing and so on. Theoretical predicted methods are considered as a rapid and cost-effective alternative for experimental evaluations. Among them, quantitative structure activity relationship (QSAR) analyses are widely used to evaluate the relationship between structures of pollutants and their toxicity value. A QSAR model can be constructed by appropriate descriptors, whether by using topological, quantum chemical, geometrical or spectral descriptors. Among them, quantum chemical descriptors are very useful, and have recently become more important because of their accuracy and reliability to characterize electronic properties of molecules (Eldred and Jurs, 1999; Zhu et al., 2014; Paukku and Hill, 2012; Chen et al., 2017).
Over the last few decades, several experimental studies about the acute toxicity of SA molecules on different test organisms have been reported in aquatic (Park and Choi, 2008; Baran et al., 2006; De Liguoro et al., 2010; Isidori et al., 2005; Białk-Bielinska et al., 2017), terrestrial enviroments (Białk-Bielinska et al., 2011) and in food samples (Hiba et al., 2016). Most of the toxicity experiments have been done in aerobic environment conditions (Zou et al., 2012), while Qin et al. (2016) have examined the toxicities of SA molecules both in aeorobic and aneorobic conditions. The chronic toxicity and the relationship between the acute and chronic toxicity of SA molecules have also been studied by several researchers (Zou et al., 2013; Long et al., 2016; Yao et al., 2013; De Liguoro et al., 2009; Bartlett et al., 2013; Wang et al., 2017; Jiang et al., 2010). However, information on sulfonamide toxicity is still insufficient, and risk assessments for these compounds need to be improved. According to our literature review, until now, toxicity studies of SAs based on quantum chemical calculations have not been reported.
In this study,
the structures of SA molecules were investigated theoretically, with the
purpose of finding exact quantum chemical descriptors for predicting toxicity
of SAs. Various quantum chemical descriptors such as the difference in energy
between frontier orbitals (![]() ), chemical
potential (
), chemical
potential (![]() ), hardness (
), hardness (![]() ), dipole
moment (
), dipole
moment (![]() ), sulfur
atom’s charge (
), sulfur
atom’s charge (![]() ) and
octanol-water partition cofficient (
) and
octanol-water partition cofficient (![]() ), were used
to develop QSAR models for the toxicity value
), were used
to develop QSAR models for the toxicity value ![]() of SA molecules.
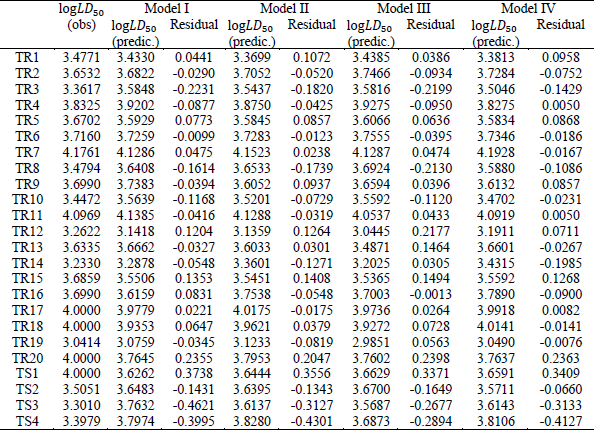
The obtained models were validated internally and externally. Among all the
calculated quantum chemical descriptors, the best ones were found via statistical
analysis.
of SA molecules.
The obtained models were validated internally and externally. Among all the
calculated quantum chemical descriptors, the best ones were found via statistical
analysis.
II. METHODS
A. Data set
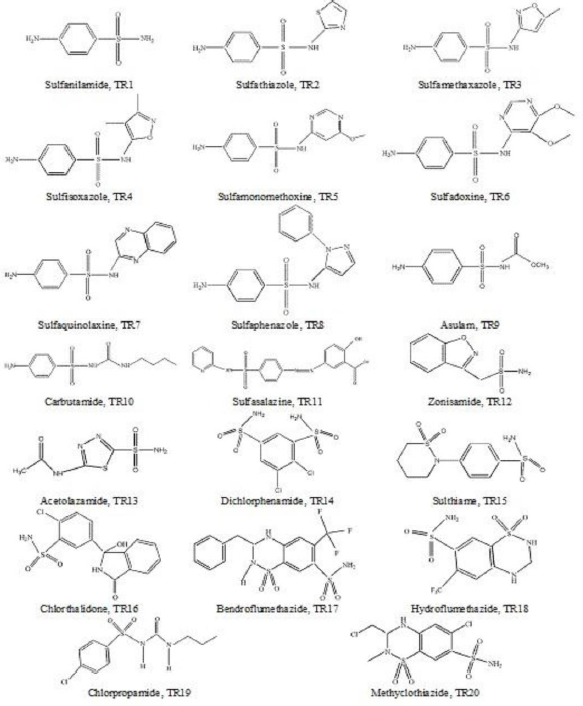
The
general structure of SAs were given in Fig. 1. In this study, the structures of
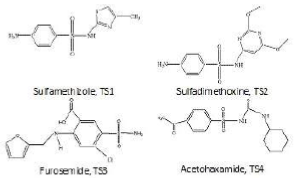
24 SA molecules were investigated theoretically. The data set for SA molecules
was divided into a training set (consisting of 20 molecules) and a test set of
4 molecules. The structures of SA molecules are shown in Fig. 2 for the
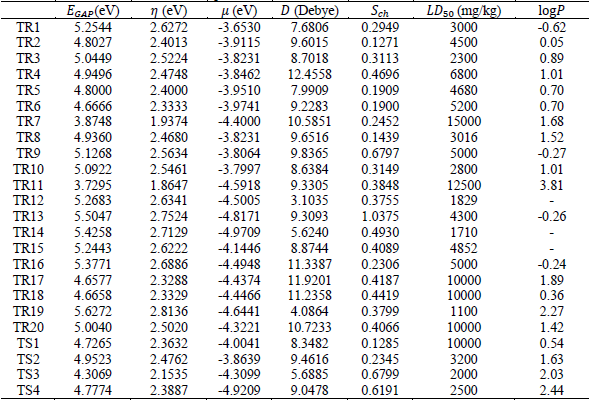
training set, and Fig. 3 for test set. Toxicity is quantified in terms of LD50, which means the amount of chemical
mg.kg-1 per body weight that causes
the death of 50 % of test animals. The oral lethal dose LD50 values for mouse oral and the octanol water coefficient, ![]() , were
obtained from the literature and are summarized in Table 1 (Toxnet, 2017).
, were
obtained from the literature and are summarized in Table 1 (Toxnet, 2017).
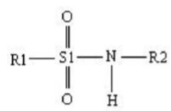
Figure 1. General structure of sulfonamides.
B. Computational Details
In this study, all computational
calculations were carried out using the Gaussian 09 software suite (Frisch et
al., 2009). The geometries of 20 SAs of the training set and 4 SAs of the
test set were optimized by density functional theory (DFT). The DFT
calculations were carried out by the hybrid B3LYP functional, which combines HF
and Becke exchange terms with the Lee–Yang–Parr correlation functional by using
6-311++G(d,p) basis set (Hehre et al., 1976). Vibrational frequency
analyzes were also calculated at the optimized geometry to ensure that the
optimized structures are at the stationary points corresponding to local minima
without any imaginary frequency. The solvation effects were computed using CPCM
as the solvation model. The solvent was water at 25°C, with dielectric constant
![]() .
.
C. Molecular Descriptors
Quantum chemical descriptors are derived within the framework of the density functional theory. These descriptors provide valuable information about reactivity of molecules. In this study, we have calculated the global descriptors such as chemical potential μ and hardness η. According to DFT, chemical potential and hardness are given by: